
 微信端
微信端

 支付宝
支付宝

 IOS版
IOS版

 安卓版
安卓版

 钉钉端
钉钉端

 客户端
客户端























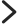
























我们e签宝已成单的医药客户可不少:
同仁堂、云南白药、恒瑞、扬子江等
药企相关的规范你要知道~
GXP 一词囊括了多种与合规性相关的活动规范,指适用于制造和开发食品以及药物、医疗设备和医疗软件应用程序等医疗产品的生命科学企业或组织的法规和准则。
看到这里,这么多的XP,他跟我们有什么关系呢?





我们e签宝已成单的医药客户可不少:
同仁堂、云南白药、恒瑞、扬子江等
药企相关的规范你要知道~
GXP 一词囊括了多种与合规性相关的活动规范,指适用于制造和开发食品以及药物、医疗设备和医疗软件应用程序等医疗产品的生命科学企业或组织的法规和准则。
看到这里,这么多的XP,他跟我们有什么关系呢?

巨大的需求!
各类管理规范中对签署的文件内容和签署有明确要求!
GXP 文件(制药行业各类管理规范)审核签字线下进行,为用户提供更优质的服务,解决当前线下文件签署多、分布广、管控效率低以及签署记录追溯难等问题,建设统一印章平台,实现内部签批文件、记录电子化。
这么棒!直接怼上去不就行啦?——项目入围、开发、对接、实施、验收等过程管理请SFR关注 GAMP 5
国际药物工程协会(ISPE)从确保计算机系统既能满足预定用途,又能符合GXP法要求出发,组织专家编写的一套简称为GAMP的 方法性 指南文件,已更新出版了5版。
按照GAMP 5药品生产自动化管理规范、GMP标准,按照4类软件对统一印章平台进行系统验证,系统要符合相关要求和标准。系统验证通过后系统才可以上线使用!
哪有那么多时间学习?
作为课代表,我帮大家整理了关键验证环节
别滑走!还有~
FDA 21 CFR
国内的药物、生物医药相关产品如果需要销售给美国相关单位和人员,那么就都应该符合21 CFR Part 11的规定。如违反,FDA能够根据规定剥夺出口到美国的权利。
21 CFR Part 11的规定是完全符合国内质量管理中关于记录控制的要求的。
也就是说如果客户的药品要销往美国,那必须使用FDA签名样式;如果是销往国内的就不需要。
差别是啥?
第11.50条要求签名展示必须包含与电子记录签名有关的信息。此信息必须包括签名人的打印姓名、签名执行的日期和时间,以及与签名相关的含义(如审查、批准、责任和作者身份)。此外,该信息受到与电子记录相同的控制,并且必须包含在人类可读的任何电子记录形式(如电子显示器或打印输出)中。
简单地说:
FAD签名样式:签名块包含手写体+印刷体签名,包含签字意见、时间戳等。
长这样:

附录:GAMP5 软件分类表


GAMP 5 软件分类表



完善信息立即免费体验!
提交成功!
我们的顾问会在1个工作日内与您取得联系
完善信息立即免费体验!




 在线客服
在线客服
 电话咨询
电话咨询